Whole Community
Prepare Data Set
The first step is to read in the whole community fish microbiome data.
ps.whole <- readRDS("rdata/p3/ps_16S_bocas_fish_final.rds")
ps.wholephyloseq-class experiment-level object
otu_table() OTU Table: [ 3113 taxa and 89 samples ]
sample_data() Sample Data: [ 89 samples by 11 sample variables ]
tax_table() Taxonomy Table: [ 3113 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 3113 tips and 3112 internal nodes ]Then get the ASV table and transpose the table so ASVs are rows. Also get the phylogenetic tree and make sure the ASV names in the ASV table and the tip names in the phylogenetic tree are identical.
asv.whole <- t(otu_table(ps.whole))
tree.whole <- phy_tree(ps.whole)
identical(sort(rownames(asv.whole)), sort(tree.whole$tip.label))[1] TRUEFinally, normalize the ASV table to relative abundance.
asv.whole.norm <- microbiome::transform(asv.whole, transform = "compositional")Alpha Diversity Estimates
Hill Diversity
hillq0 <- estimate_richness(ps.whole, measures = "Observed")Shannon Exponential
shannon.hill <- exp(vegan::diversity(t(asv.whole.norm), index = "shannon"))
shannon.whole.df <- as.data.frame(shannon.hill)Simpson Index
1/(1-(vegan::diversity(t(asv.whole.norm), index = "simpson")))
simpson.hill<-1/(1-(vegan::diversity(t(asv.whole.norm), index = "simpson")))
simpson.whole.df <- as.data.frame(simpson.hill)Now we combine the phyloseq object, sample data, and add new columns with Hill diversity into one data frame and save the object.
newDF.whole <- data.frame(hillq0, shannon.hill, simpson.hill,
sample_data(ps.whole))
ps.whole.hill <- merge_phyloseq(otu_table(ps.whole),
sample_data(newDF.whole),
tax_table(ps.whole),
phy_tree(ps.whole))
ps.whole.hill
sample_data(ps.whole.hill)
saveRDS(ps.whole.hill, "rdata/p4/ps_16S_bocas_fish_final_hill.rds")phyloseq-class experiment-level object
otu_table() OTU Table: [ 3113 taxa and 89 samples ]
sample_data() Sample Data: [ 89 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 3113 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 3113 tips and 3112 internal nodes ]Alpha Diversity Estimates by Zone
Next, we calculate alpha diversity with Hill numbers for each zone using alpha_div() function.
ps.outer <- subset_samples(ps.whole.hill, Zone == "Outer bay")
ps.outer <- prune_taxa(taxa_sums(ps.outer) > 0, ps.outer)
ps.outer
ps.inner <- subset_samples(ps.whole.hill, Zone == "Inner bay")
ps.inner <- prune_taxa(taxa_sums(ps.inner) > 0, ps.inner)
ps.inner
ps.inner.dist <- subset_samples(ps.whole.hill, Zone == "Inner bay disturbed")
ps.inner.dist <- prune_taxa(taxa_sums(ps.inner.dist) > 0, ps.inner.dist)
ps.inner.distphyloseq-class experiment-level object
otu_table() OTU Table: [ 1271 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 1271 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 1271 tips and 1270 internal nodes ]phyloseq-class experiment-level object
otu_table() OTU Table: [ 1590 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 1590 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 1590 tips and 1589 internal nodes ]phyloseq-class experiment-level object
otu_table() OTU Table: [ 909 taxa and 19 samples ]
sample_data() Sample Data: [ 19 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 909 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 909 tips and 908 internal nodes ]Get the ASV tables and transpose the tables so ASVs are rows.
asv.outer <- t(otu_table(ps.outer))
asv.inner <- t(otu_table(ps.inner))
asv.inner.dist <- t(otu_table(ps.inner.dist))And transform the counts to relative abundance.
asv.outer.norm <- microbiome::transform(asv.outer, transform = "compositional")
asv.inner.norm <- microbiome::transform(asv.inner, transform = "compositional")
asv.inner.dist.norm <- microbiome::transform(asv.inner.dist,
transform = "compositional")Now we can test alpha diversity per zone based on Hill numbers at different diversity levels (q = 0 (observed), q = 1 (shannon exponential), q = 2 (multiplicative simpson)).
Note: for Hill numbers, alpha diversity per zone cannot be obtained as a mean across samples. The calculation is different as performed by function alpha_div().
alpha_div(countable = asv.outer.norm, qvalue = 0)
alpha_div(countable = asv.outer.norm, qvalue = 1)
alpha_div(countable = asv.outer.norm, qvalue = 2)
alpha_div(countable = asv.inner.norm, qvalue = 0)
alpha_div(countable = asv.inner.norm, qvalue = 1)
alpha_div(countable = asv.inner.norm, qvalue = 2)
alpha_div(countable = asv.inner.dist.norm, qvalue = 0)
alpha_div(countable = asv.inner.dist.norm, qvalue = 1)
alpha_div(countable = asv.inner.dist.norm, qvalue = 2)Significance Test
Here we run Kruskal-Wallis Tests on each order of diversty (q value) and posthoc Dunn Tests with Benjamin Hochberg correction for zone comparison.
temp_table <- data.frame(sample_data(ps.whole))
temp_table[, c(1:4, 7:11)] <- list(NULL)
temp_table <- temp_table %>% tibble::rownames_to_column("Sample")
temp_table <- temp_table %>% dplyr::rename(Group = Zone)
temp_table$Group <- as.character(temp_table$Group)
temp_table$Reef <- as.character(temp_table$Reef)
hill_hierarchy <- temp_table[,1:2]
hill_hierarchy2 <- temp_table[, c(1, 3)]By Zone
divtestresult.q0 <- div_test(asv.whole.norm, qvalue = 0,
hierarchy = hill_hierarchy, posthoc = TRUE)
divtestresult.q1 <- div_test(asv.whole.norm, qvalue = 1,
hierarchy = hill_hierarchy, posthoc = TRUE)
divtestresult.q2 <- div_test(asv.whole.norm, qvalue = 2,
hierarchy = hill_hierarchy, posthoc = TRUE)| q-value | homogeneity.pvalue | normality.pvalue | method | posthoc.method |
|---|---|---|---|---|
| 0 | 0.4515383 | 0 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 1 | 0.2115412 | 0 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 2 | 0.016806 | 0 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
q-value = 0
Z P.unadj P.adj
Inner bay-Inner bay disturbed 0.008682602 0.99307237 0.9930724
Inner bay-Outer bay 1.700501330 0.08903667 0.2671100
Inner bay disturbed-Outer bay 1.417817787 0.15624397 0.2343660q-value = 1
Z P.unadj P.adj
Inner bay-Inner bay disturbed -1.416080 0.156752013 0.156752013
Inner bay-Outer bay 2.128057 0.033332390 0.049998585
Inner bay disturbed-Outer bay 3.201244 0.001368355 0.004105066q-value = 2
Z P.unadj P.adj
Inner bay-Inner bay disturbed -1.579482 0.114225549 0.1142255
Inner bay-Outer bay 1.586790 0.112560206 0.1688403
Inner bay disturbed-Outer bay 2.910593 0.003607432 0.0108223By Reef
divtestresult.q0 <- div_test(asv.whole.norm, qvalue = 0,
hierarchy = hill_hierarchy2, posthoc = TRUE)
divtestresult.q1 <- div_test(asv.whole.norm, qvalue = 1,
hierarchy = hill_hierarchy2, posthoc = TRUE)
divtestresult.q2 <- div_test(asv.whole.norm, qvalue = 2,
hierarchy = hill_hierarchy2, posthoc = TRUE)| q-value | homogeneity.pvalue | normality.pvalue | method | posthoc.method |
|---|---|---|---|---|
| 0 | 0.0027403 | 0 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 1 | 0.0003942 | 0 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 2 | 0.0000162 | 0 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
q-value = 0
q-value = 1
q-value = 2
data.hill.whole_n <- data.frame(sample_data(ps.whole.hill)) %>%
pivot_longer(cols = 1:3, names_to = "Index", values_to = "Diversity")
data.hill.whole_n$Fraction_delailed <- NULL
data.hill.whole_n <- data.hill.whole_n[, c(10,1,2,3,4,5,6,7,8,9,12,11)]
data.hill.whole_n <- dplyr::mutate_if(data.hill.whole_n, is.factor, as.character)
data.hill.whole_n$Index <- stringr::str_replace(
data.hill.whole_n$Index, "Observed", "Observed (q=0)")
data.hill.whole_n$Index <- stringr::str_replace(
data.hill.whole_n$Index, "shannon.hill", "Shannon exponential (q=1)")
data.hill.whole_n$Index <- stringr::str_replace(
data.hill.whole_n$Index, "simpson.hill", "Simpson multiplicative inverse (q=2)")
data.hill.whole_n <- data.hill.whole_n[order(data.hill.whole_n$Index),]Core Community
This code is basically the same as the code above, but this time on the core community identified using the Indicator analysis described in the first script.
Prepare Data Set
The first step is to read in the whole community fish microbiome data.
ps.core <- readRDS("rdata/p1/ps_indv01_core_fish.rds")
ps.corephyloseq-class experiment-level object
otu_table() OTU Table: [ 27 taxa and 89 samples ]
sample_data() Sample Data: [ 89 samples by 11 sample variables ]
tax_table() Taxonomy Table: [ 27 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 27 tips and 26 internal nodes ]Then get the ASV table and transpose the table so ASVs are rows. Also get the phylogenetic tree and make sure the ASV names in the ASV table and the tip names in the phylogenetic tree are identical.
asv.core <- t(otu_table(ps.core))
tree.core <- phy_tree(ps.core)
identical(sort(rownames(asv.core)), sort(tree.core$tip.label))[1] TRUEFinally, normalize the ASV table to relative abundance.
asv.core.norm <- microbiome::transform(asv.core, transform = "compositional")Alpha Diversity Estimates
Hill Diversity
hillq0 <- estimate_richness(ps.core, measures = "Observed")Shannon Exponential
shannon.hill <- exp(vegan::diversity(t(asv.core.norm), index = "shannon"))
shannon.core.df <- as.data.frame(shannon.hill)Simpson Index
1/(1-(vegan::diversity(t(asv.core.norm), index = "simpson")))
simpson.hill <- 1/(1-(vegan::diversity(t(asv.core.norm), index = "simpson")))
simpson.core.df <- as.data.frame(simpson.hill)Now we combine the phyloseq object, sample data, and add new columns with Hill diversity into one data frame and save the object.
newDF.core <- data.frame(hillq0, shannon.hill,
simpson.hill, sample_data(ps.core))
ps.core.hill <- merge_phyloseq(otu_table(ps.core),
sample_data(newDF.core),
tax_table(ps.core),
phy_tree(ps.core))
saveRDS(ps.core.hill, "rdata/p4/ps_core_hill.rds")phyloseq-class experiment-level object
otu_table() OTU Table: [ 27 taxa and 89 samples ]
sample_data() Sample Data: [ 89 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 27 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 27 tips and 26 internal nodes ]Alpha Diversity Estimates by Zone
Next, we calculate alpha diversity with Hill numbers for each zone using alpha_div() function.
ps.outer <- subset_samples(ps.core.hill, Zone == "Outer bay")
ps.outer <- prune_taxa(taxa_sums(ps.outer) > 0, ps.outer)
ps.outer
ps.inner <- subset_samples(ps.core.hill, Zone == "Inner bay")
ps.inner <- prune_taxa(taxa_sums(ps.inner) > 0, ps.inner)
ps.inner
ps.inner.dist <- subset_samples(ps.core.hill, Zone == "Inner bay disturbed")
ps.inner.dist <- prune_taxa(taxa_sums(ps.inner.dist) > 0, ps.inner.dist)
ps.inner.distphyloseq-class experiment-level object
otu_table() OTU Table: [ 27 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 27 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 27 tips and 26 internal nodes ]phyloseq-class experiment-level object
otu_table() OTU Table: [ 27 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 27 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 27 tips and 26 internal nodes ]phyloseq-class experiment-level object
otu_table() OTU Table: [ 27 taxa and 19 samples ]
sample_data() Sample Data: [ 19 samples by 14 sample variables ]
tax_table() Taxonomy Table: [ 27 taxa by 11 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 27 tips and 26 internal nodes ]Get the ASV tables and transpose the tables so ASVs are rows.
asv.outer <- t(otu_table(ps.outer))
asv.inner <- t(otu_table(ps.inner))
asv.inner.dist <- t(otu_table(ps.inner.dist))And transform the counts to relative abundance.
asv.outer.norm <- microbiome::transform(asv.outer, transform = "compositional")
asv.inner.norm <- microbiome::transform(asv.inner, transform = "compositional")
asv.inner.dist.norm <- microbiome::transform(asv.inner.dist, transform = "compositional")Now we can test alpha diversity per zone based on Hill numbers at different diversity levels (q = 0 (observed), q = 1 (shannon exponential), q = 2 (multiplicative simpson)).
Note: for Hill numbers, alpha diversity per zone cannot be obtained as a mean across samples. The calculation is different as performed by function alpha_div().
alpha_div(countable = asv.outer.norm, qvalue = 0)
alpha_div(countable = asv.outer.norm, qvalue = 1)
alpha_div(countable = asv.outer.norm, qvalue = 2)
alpha_div(countable = asv.inner.norm, qvalue = 0)
alpha_div(countable = asv.inner.norm, qvalue = 1)
alpha_div(countable = asv.inner.norm, qvalue = 2)
alpha_div(countable = asv.inner.dist.norm, qvalue = 0)
alpha_div(countable = asv.inner.dist.norm, qvalue = 1)
alpha_div(countable = asv.inner.dist.norm, qvalue = 2)Significance Test
Here we run Kruskal-Wallis Tests on each order of diversity (q value) and posthoc Dunn Tests with Benjamin Hochberg correction for zone comparison.
temp_table <- data.frame(sample_data(ps.core))
temp_table[, c(1:4, 7:11)] <- list(NULL)
temp_table <- temp_table %>% tibble::rownames_to_column("Sample")
temp_table <- temp_table %>% dplyr::rename(Group = Zone)
temp_table$Group <- as.character(temp_table$Group)
temp_table$Reef <- as.character(temp_table$Reef)
hill_hierarchy <- temp_table[,1:2]
hill_hierarchy2 <- temp_table[, c(1, 3)]By Zone
divtestresult.q1 <- div_test(asv.core.norm, qvalue = 1,
hierarchy = hill_hierarchy, posthoc = TRUE)
divtestresult.q2 <- div_test(asv.core.norm, qvalue = 2,
hierarchy = hill_hierarchy, posthoc = TRUE)| q-value | homogeneity.pvalue | normality.pvalue | method | posthoc.method |
|---|---|---|---|---|
| 1 | 0.6000394 | 0.0000041 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 2 | 0.3376986 | 0.0000004 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
q-value = 1
Z P.unadj P.adj
Inner bay-Inner bay disturbed -1.633200 0.102426879 0.15364032
Inner bay-Outer bay 1.480387 0.138769942 0.13876994
Inner bay disturbed-Outer bay 2.875053 0.004039591 0.01211877q-value = 2
Z P.unadj P.adj
Inner bay-Inner bay disturbed -1.944889 0.051788329 0.07768249
Inner bay-Outer bay 1.105664 0.268871893 0.26887189
Inner bay disturbed-Outer bay 2.872398 0.004073694 0.01222108By Reef
divtestresult.q1 <- div_test(asv.core.norm, qvalue = 1,
hierarchy = hill_hierarchy2, posthoc = TRUE)
divtestresult.q2 <- div_test(asv.core.norm, qvalue = 2,
hierarchy = hill_hierarchy2, posthoc = TRUE)| q-value | homogeneity.pvalue | normality.pvalue | method | posthoc.method |
|---|---|---|---|---|
| 1 | 0.0003462 | 0.0000041 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 2 | 0.0000402 | 0.0000004 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
q-value = 1
q-value = 2
hill.core.DF <- data.frame(sample_data(ps.core.hill))
kruskal.test(Observed~Zone, data = hill.core.DF)
kruskal.test(Observed~Reef, data = hill.core.DF)
dunn.core.obs.zone <- dunn.test(hill.core.DF$Observed, hill.core.DF$Zone,
method="hochberg", table=TRUE, wrap=TRUE)
dunn.core.obs.reef <- dunn.test(hill.core.DF$Observed, hill.core.DF$Reef,
method="hochberg", table = TRUE, wrap = TRUE)
dunn.test(hill.core.DF$Observed, hill.core.DF$Zone, method="none")
dunn.test(hill.core.DF$Observed, hill.core.DF$Reef, method="none")| q-value | homogeneity.pvalue | normality.pvalue | method | posthoc.method |
|---|---|---|---|---|
| 1 | 0.0003462 | 0.0000041 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
| 2 | 0.0000402 | 0.0000004 | Kruskal-Wallis Test | Dunn test with Benjamini-Hochberg correction |
q-value = 1
q-value = 2
data.hill.core_n <- hill.core.DF %>%
pivot_longer(cols = 1:3, names_to = "Index", values_to = "Diversity")
data.hill.core_n$Fraction_delailed <- NULL
data.hill.core_n <- data.hill.core_n[, c(10,1,2,3,4,5,6,7,8,9,12,11)]
data.hill.core_n <- dplyr::mutate_if(data.hill.core_n, is.factor, as.character)
data.hill.core_n$Index <- stringr::str_replace(
data.hill.core_n$Index, "Observed", "Observed (q=0)")
data.hill.core_n$Index <- stringr::str_replace(
data.hill.core_n$Index, "shannon.hill", "Shannon exponential (q=1)")
data.hill.core_n$Index <- stringr::str_replace(
data.hill.core_n$Index, "simpson.hill", "Simpson multiplicative inverse (q=2)")
data.hill.core_n <- data.hill.core_n[order(data.hill.core_n$Index),]Analysis Summary
Summary Table
data.hill.whole <- readRDS("rdata/p4/data.hill.whole_n.rds")
data.hill.core <- readRDS("rdata/p4/data.hill.core_n.rds")Summary Figure
#, foldcode=TRUE
level_order3 <- c('SCR', 'PPR', 'CCR', 'ALR', 'SIS', 'ROL', 'RNW', 'PST','PBL')
level_order <- c('Outer bay', 'Inner bay', 'Inner bay disturbed')
level_order2 <- c('Observed', 'shannon.hill', 'simpson.hill')
disp.pal4 <- c("royalblue4", "royalblue3", "royalblue1", "aquamarine4",
"mediumaquamarine", "aquamarine2", "chocolate3", "chocolate2",
"sienna1","royalblue4", "royalblue3", "royalblue1",
"aquamarine4", "mediumaquamarine", "aquamarine2", "chocolate3",
"chocolate2", "sienna1")
disp.pal5 <- c("midnightblue", "royalblue4", "royalblue3","darkslategrey",
"aquamarine4", "mediumaquamarine", "chocolate4", "sienna",
"chocolate3","midnightblue", "royalblue4", "royalblue3",
"darkslategrey","aquamarine4", "mediumaquamarine", "chocolate4",
"sienna", "chocolate3")data.hill.whole$Reef <- factor(data.hill.whole$Reef,
levels = c('SCR', 'PPR', 'CCR', 'ALR', 'SIS',
'ROL', 'RNW', 'PST','PBL'))
data.hill.core$Reef <- factor(data.hill.core$Reef,
levels = c('SCR', 'PPR', 'CCR', 'ALR', 'SIS',
'ROL', 'RNW', 'PST','PBL'))font_theme = theme(
axis.title.x = element_text(size = 14),
axis.text.x = element_text(size = 10),
axis.title.y = element_text(size = 14),
axis.text.y = element_text(size = 8))fig01 <-
ggplot(data = data.hill.whole, aes(x = factor(Zone, level = level_order),
y=Diversity)) +
stat_boxplot(aes(colour=Reef), geom ='errorbar') + #alpha=0.3
geom_boxplot(aes(fill=Reef, colour=Reef), alpha=0.6,
outlier.alpha = 0.7, outlier.shape = 16) +
scale_colour_manual(values=disp.pal5)+
scale_fill_manual(values=disp.pal4)+
ggtitle("Alpha Diversity", subtitle="Whole microbiome")+
theme(plot.title=element_text(size=18)) +
theme(plot.subtitle=element_text(size=16)) +
font_theme+
stat_summary(aes(fill=Reef), fun="mean", geom="point", size=2.5, shape=18,
position=position_dodge(width=0.75),
color=c( "midnightblue","royalblue4", "royalblue3",
"darkslategrey","aquamarine4","mediumaquamarine",
"chocolate4", "sienna", "chocolate3",
"midnightblue","royalblue4", "royalblue3",
"darkslategrey","aquamarine4","mediumaquamarine",
"chocolate4", "sienna", "chocolate3",
"midnightblue","royalblue4", "royalblue3",
"darkslategrey","aquamarine4","mediumaquamarine",
"chocolate4", "sienna", "chocolate3"),
show.legend = FALSE) +
scale_x_discrete(name="Zone") + scale_y_continuous(name="Hill diversity") +
theme(plot.title = element_text(family=NA, face="bold", size=16)) +
facet_wrap(~factor(Index, levels=unique(Index)),scales="free",
nrow=2, ncol = 3) +
theme(strip.background = element_rect(color="black", fill="white",
size=1, linetype="blank"),
strip.text.x = element_text(size = 14),
panel.border = element_rect(fill = NA, color="black"),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_rect(fill = "white",colour = NA),
legend.key = element_rect(fill = "transparent", color = NA),
legend.text = element_text(size = 12),
legend.title = element_text(size = 14))fig02 <-
ggplot(data = data.hill.core, aes(x = factor(Zone, level = level_order),
y=Diversity)) +
stat_boxplot(aes(colour=Reef), geom ='errorbar') +
geom_boxplot(aes(fill=Reef, colour=Reef), alpha=0.6,
outlier.alpha = 0.7, outlier.shape = 16) +
scale_colour_manual(values=disp.pal5) +
scale_fill_manual(values=disp.pal4) +
ggtitle("", subtitle="Core microbiome")+
theme(plot.subtitle = element_text(size=16))+
font_theme+
stat_summary(aes(fill=Reef), fun="mean", geom="point", size=2.5, shape=18,
position=position_dodge(width=0.75),
color=c( "midnightblue","royalblue4", "royalblue3",
"darkslategrey","aquamarine4","mediumaquamarine",
"chocolate4", "sienna", "chocolate3",
"midnightblue","royalblue4", "royalblue3",
"darkslategrey","aquamarine4","mediumaquamarine",
"chocolate4", "sienna", "chocolate3",
"midnightblue","royalblue4", "royalblue3",
"darkslategrey","aquamarine4","mediumaquamarine",
"chocolate4", "sienna", "chocolate3"),
show.legend = FALSE)+
scale_x_discrete(name="Zone") + scale_y_continuous(name="Hill diversity")+
theme(plot.title = element_text(family=NA, face="bold", size=16)) +
facet_wrap(~factor(Index, levels=unique(Index)), scales="free",
nrow=2, ncol = 3) +
theme(strip.background = element_rect(color="black", fill="white",
size=1, linetype="blank"),
strip.text.x = element_text(size = 14),
panel.border = element_rect(fill = NA, color="black"),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_rect(fill = "white",colour = NA),
legend.key = element_rect(fill = "transparent", color = NA),
legend.text = element_text(size = 12),
legend.title = element_text(size = 14))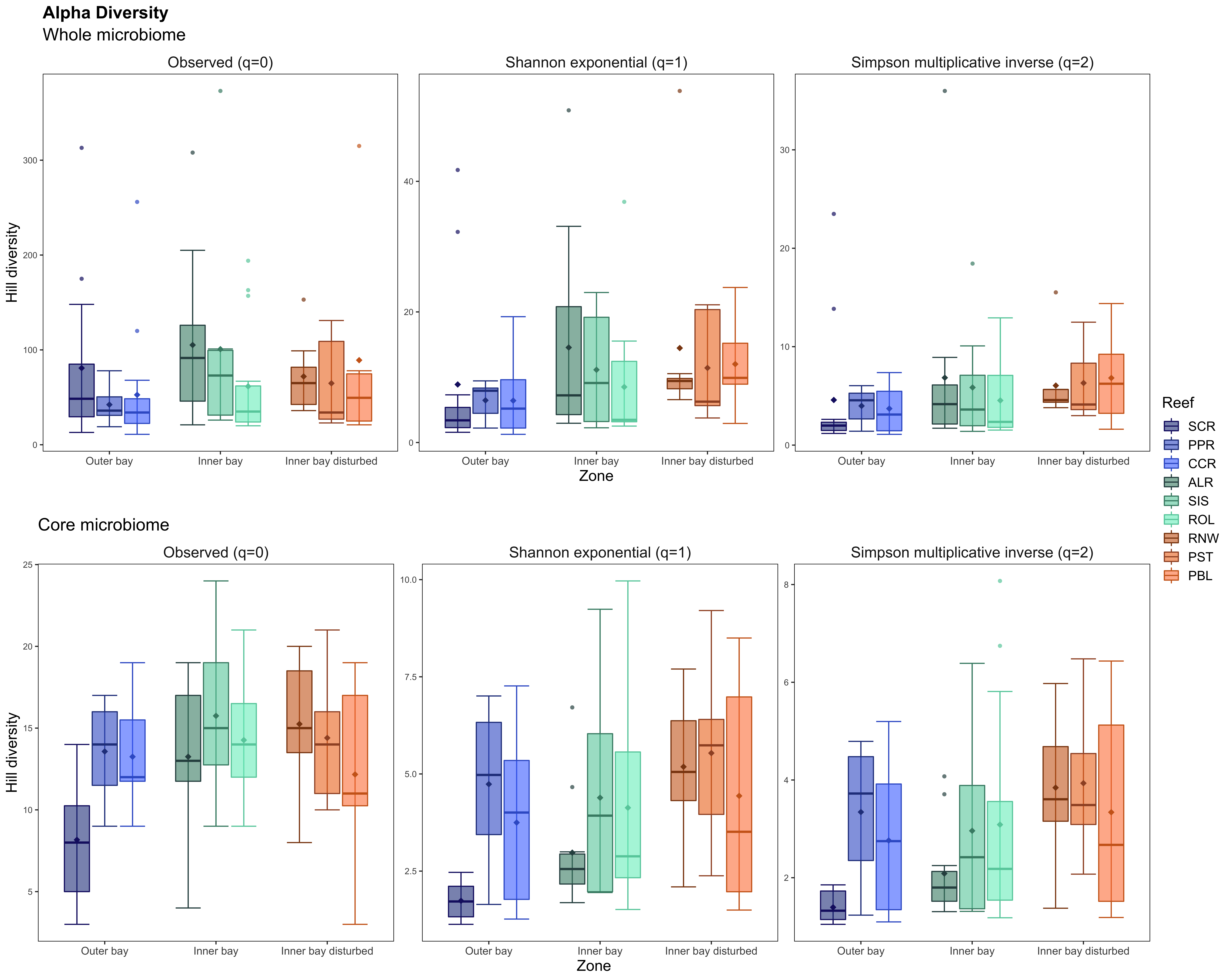
That’s the end of Script 4. In the next Script we calculate beta diversity estimates of the fish gut microbiome.
Source Code
The source code for this page can be accessed on GitHub by clicking this link.